Introduction
Cholesterol is an extremely important biological
molecule that has roles in membrane structure as
well as being a precursor for the synthesis of the
steroid hormones and bile acids . Both dietary
cholesterol and that synthesized de novo are
transported through the circulation in lipoprotein
particles . The same is true of cholesteryl esters,
the form in which cholesterol is stored in cells.
The synthesis and utilization of cholesterol must be
tightly regulated in order to prevent over-
accumulation and abnormal deposition within the
body. Of particular importance clinically is the
abnormal deposition of cholesterol and cholesterol-
rich lipoproteins in the coronary arteries. Such
deposition, eventually leading to atherosclerosis, is
the leading contributory factor in diseases of the
coronary arteries.
Biosynthesis of cholesterol
Slightly less than half of the cholesterol in the body
derives from biosynthesis de novo . Biosynthesis in
the liver accounts for approximately 10%, and in the
intestines approximately 15%, of the amount
produced each day. Cholesterol synthesis occurs in
the cytoplasm and microsomes (ER) from the two-
carbon acetate group of acetyl-CoA.
The acetyl-CoA utilized for cholesterol biosynthesis
is derived from an oxidation reaction (e.g., fatty
acids or pyruvate) in the mitochondria and is
transported to the cytoplasm by the same process
as that described for fatty acid synthesis (see the
Figure below). Acetyl-CoA can also be synthesized
from cytosolic acetate derived from cytoplasmic
oxidation of ethanol which is initiated by
cytoplasmic alcohol dehydrogenase (ADH3). All the
reduction reactions of cholesterol biosynthesis use
NADPH as a cofactor. The isoprenoid intermediates
of cholesterol biosynthesis can be diverted to other
synthesis reactions, such as those for dolichol
(used in the synthesis of N -linked glycoproteins,
coenzyme Q (of the oxidative phosphorylation
pathway) or the side chain of heme- a. Additionally,
these intermediates are used in the lipid
modification of some proteins .
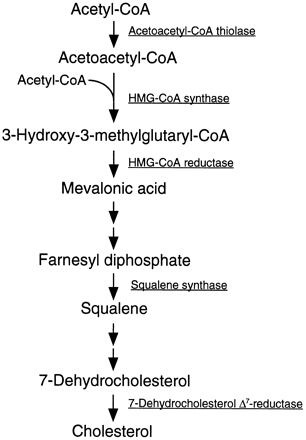
The process of cholesterol synthesis has five major
steps:
1. Acetyl-CoAs are converted to 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA)
2. HMG-CoA is converted to mevalonate
3. Mevalonate is converted to the isoprene based
molecule, isopentenyl pyrophosphate (IPP), with the concomitant loss of CO2
4. IPP is converted to squalene
5. Squalene is converted to cholesterol.
Pathway of cholesterol biosynthesis. Synthesis
begins with the transport of acetyl-CoA from the
mitochondrion to the cytosol. The rate limiting step
occurs at the 3-hydroxy-3-methylglutaryl-CoA
(HMG-CoA) reducatase, HMGR catalyzed step. The
phosphorylation reactions are required to solubilize
the isoprenoid intermediates in the pathway.
Intermediates in the pathway are used for the
synthesis of prenylated proteins, dolichol, coenzyme
Q and the side chain of heme a . The abbreviation
"PP" (e.g. isopentenyl-PP) stands for
pyrophosphate. Place mouse over intermediate
names to see structure.
Acetyl-CoA units are converted to mevalonate by a
series of reactions that begins with the formation of
HMG-CoA . Unlike the HMG-CoA formed during
ketone body synthesis in the mitochondria, this
form is synthesized in the cytoplasm. However, the
pathway and the necessary enzymes are similar to
those in the mitochondria. Two moles of acetyl-CoA
are condensed in a reversal of the thiolase reaction,
forming acetoacetyl-CoA. The cytoplasmic thiolase
enzyme involved in cholesterol biosynthesis is
acetoacetyl-CoA thiolase encoded by the ACAT2
gene. Although the bulk of acetoacetyl-CoA is
derived via this process, it is possible for some
acetoacetate, generated during ketogenesis, to
diffuse out of the mitochondria and be converted to
acetoacetyl-CoA in the cytosol via the action of
acetoacetyl-CoA synthetase (AACS). Acetoacetyl-
CoA and a third mole of acetyl-CoA are converted to
HMG-CoA by the action of HMG-CoA synthase.
HMG-CoA is converted to mevalonate by HMG-CoA
reductase, HMGR (this enzyme is bound in the
endoplasmic reticulum, ER). HMGR absolutely
requires NADPH as a cofactor and two moles of
NADPH are consumed during the conversion of
HMG-CoA to mevalonate. The reaction catalyzed by
HMGR is the rate limiting step of cholesterol
biosynthesis, and this enzyme is subject to
complex regulatory controls as discussed below.
Mevalonate is then activated by two successive
phosphorylations (catalyzed by mevalonate kinase,
and phosphomevalonate kinase), yielding 5-
pyrophosphomevalonate. In humans, mevalonate
kinase resides in the cytosol indicating that not all
the reactions of cholesterol synthesis are catalyzed
by membrane-associated enzymes as originally
described. After phosphorylation, an ATP-dependent
decarboxylation yields isopentenyl pyrophosphate,
IPP, an activated isoprenoid molecule. Isopentenyl
pyrophosphate is in equilibrium with its isomer,
dimethylallyl pyrophosphate, DMPP. One molecule
of IPP condenses with one molecule of DMPP to
generate geranyl pyrophosphate, GPP. GPP further
condenses with another IPP molecule to yield
farnesyl pyrophosphate, FPP. Finally, the NADPH-
requiring enzyme, squalene synthase catalyzes the
head-to-tail condensation of two molecules of FPP,
yielding squalene. Like HMGR, squalene synthase is
tightly associated with the ER. Squalene undergoes
a two step cyclization to yield lanosterol. The first
reaction is catalyzed by squalene monooxygenase.
This enzyme uses NADPH as a cofactor to
introduce molecular oxygen as an epoxide at the 2,3
position of squalene. Through a series of 19
additional reactions, lanosterol is converted to
cholesterol.
The terminal reaction in cholesterol biosynthesis is
catalyzed by the enzyme 7-dehydrocholesterol
reductase encoded by the DHCR7 gene. Functional
DHCR7 protein is a 55.5 kDa NADPH-requiring integral membrane protein localized to the microsomal membrane. Deficiency in DHCR7 (due to gene mutations) results in the disorder called Smith-Lemli-Opitz syndrome, SLOS . SLOS is characterized by increased levels of 7-dehydrocholesterol and reduced levels (15% to 27% of normal) of cholesterol resulting in multiple developmental malformations and behavioral problems.
Regulation of cholesterol synthesis
Normal healthy adults synthesize cholesterol at a
rate of approximately 1g/day and consume
approximately 0.3g/day. A relatively constant level
of cholesterol in the blood (150–200 mg/dL) is
maintained primarily by controlling the level of de
novo synthesis. The level of cholesterol synthesis is
regulated in part by the dietary intake of cholesterol.
Cholesterol from both diet and synthesis is utilized
in the formation of membranes and in the synthesis of the steroid hormones and bile acids . The greatest proportion of cholesterol is used in bile acid synthesis.
The cellular supply of cholesterol is maintained at a
steady level by three distinct mechanisms:
1. Regulation of HMGR activity and levels
2. Regulation of excess intracellular free cholesterol
through the activity of acyl-CoA:cholesterol
acyltransferase, ACAT
3. Regulation of plasma cholesterol levels via LDL receptor-mediated uptake and HDL-mediated reverse transport.
Regulation of HMGR activity is the primary means
for controlling the level of cholesterol biosynthesis.
The enzyme is controlled by four distinct
mechanisms: feed-back inhibition, control of gene
expression, rate of enzyme degradation and
phosphorylation-dephosphorylation.
The first three control mechanisms are exerted by
cholesterol itself. Cholesterol acts as a feed-back
inhibitor of pre-existing HMGR as well as inducing
rapid degradation of the enzyme. The latter is the
result of cholesterol-induced polyubiquitination of
HMGR and its degradation in the proteosome (see
proteolytic degradation below). This ability of
cholesterol is a consequence of the sterol sensing
domain, SSD of HMGR. In addition, when cholesterol
is in excess the amount of mRNA for HMGR is
reduced as a result of decreased expression of the
gene. The mechanism by which cholesterol (and
other sterols) affect the transcription of the HMGR
gene is described below under regulation of sterol
content .
Regulation of HMGR through covalent modification
occurs as a result of phosphorylation and
dephosphorylation. The enzyme is most active in its
unmodified form. Phosphorylation of the enzyme
decreases its activity. HMGR is phosphorylated by
AMP-activated protein kinase, AMPK (this is not the
same as cAMP-dependent protein kinase, PKA).
AMPK itself is activated via phosphorylation.
Phosphorylation of AMPK is catalyzed by at least 2
enzymes. The primary kinase sensitive to rising
AMP levels is LKB1. LKB1 was first identified as a
gene in humans carrying an autosomal dominant
mutation in Peutz-Jeghers syndrome, PJS. LKB1 is
also found mutated in lung adenocarcinomas. The
second AMPK phosphorylating enzyme is
calmodulin-dependent protein kinase kinase-beta
(CaMKKβ). CaMKKβ induces phosphorylation of
AMPK in response to increases in intracellular Ca2+
as a result of muscle contraction. Visit AMPK: The
Master Metabolic Regulator for more detailed
information on the role of AMPK in regulating
metabolism.
Regulation of HMGR by covalent modification. HMGR
is most active in the dephosphorylated state.
Phosphorylation is catalyzed by AMP-activated
protein kinase (AMPK) an enzyme whose activity is
also regulated by phosphorylation. Phosphorylation
of AMPK is catalyzed by at least 2 enzymes: LKB1
and CaMKKβ. Hormones such as glucagon and
epinephrine negatively affect cholesterol
biosynthesis by increasing the activity of the
inhibitor of phosphoprotein phosphatase inhibitor-1,
PPI-1. Conversely, insulin stimulates the removal of
phosphates and, thereby, activates HMGR activity.
Additional regulation of HMGR occurs through an
inhibition of its' activity as well as of its' synthesis
by elevation in intracellular cholesterol levels. This
latter phenomenon involves the transcription factor
SREBP described below.
The activity of HMGR is additionally controlled by
the cAMP signaling pathway. Increases in cAMP
lead to activation of cAMP-dependent protein
kinase, PKA. In the context of HMGR regulation, PKA
phosphorylates phosphoprotein phosphatase
inhibitor-1 (PPI-1) leading to an increase in its'
activity. PPI-1 can inhibit the activity of numerous
phosphatases including protein phosphatase 2C
(PP2C) and PP2A (also called HMGR phosphatase)
which remove phosphates from AMPK and HMGR,
respectively. This maintains AMPK in the
phosphorylated and active state, and HMGR in the
phosphorylated and inactive state. As the stimulus
leading to increased cAMP production is removed,
the level of phosphorylations decreases and that of
dephosphorylations increases. The net result is a
return to a higher level of HMGR activity.
Since the intracellular level of cAMP is regulated by
hormonal stimuli, regulation of cholesterol
biosynthesis is hormonally controlled. Insulin leads
to a decrease in cAMP, which in turn activates
cholesterol synthesis. Alternatively, glucagon and
epinephrine, which increase the level of cAMP,
inhibit cholesterol synthesis.
The ability of insulin to stimulate, and glucagon to
inhibit, HMGR activity is consistent with the effects
of these hormones on other metabolic pathways.
The basic function of these two hormones is to
control the availability and delivery of energy to all
cells of the body.
Long-term control of HMGR activity is exerted
primarily through control over the synthesis and
degradation of the enzyme. When levels of
cholesterol are high, the level of expression of the
HMGR gene is reduced. Conversely, reduced levels of cholesterol activate expression of the gene. Insulin also brings about long-term regulation of cholesterol metabolism by increasing the level of HMGR synthesis.





No comments:
Write comments